AutoLabs: A Multi-Agent System for Autonomous Chemical Experimentation
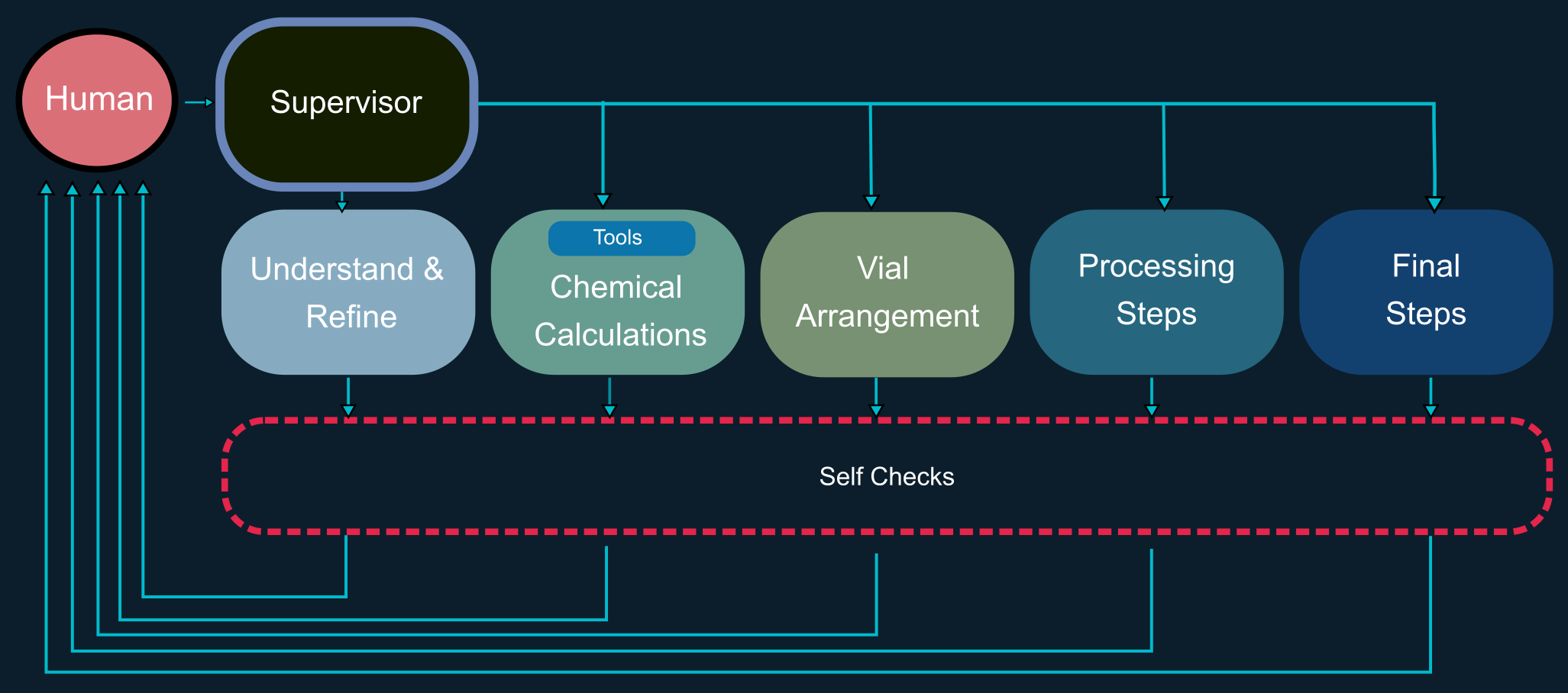
AutoLabs, built with LangGraph, has been demonstrated to generate accurate hardware files for executing experiments on the Big Kahuna robotic platform from Unchained Labs. Analysis shows that the quantitative accuracy of outputs is primarily driven by the reasoning capacity of the underlying LLM. A multi-agent architecture that decomposes tasks across specialized agents produces more robust and effective protocol generation. Built-in self-correction mechanisms improve both procedural and numerical accuracy. The value of external computational tools depends on the agent’s ability to use them reliably. Agents with higher reasoning capacity reach solutions more directly, following shorter and more computationally efficient pathways.
FragNet: A Graph Neural Network for Molecular Property Prediction with Four Layers of Interpretability
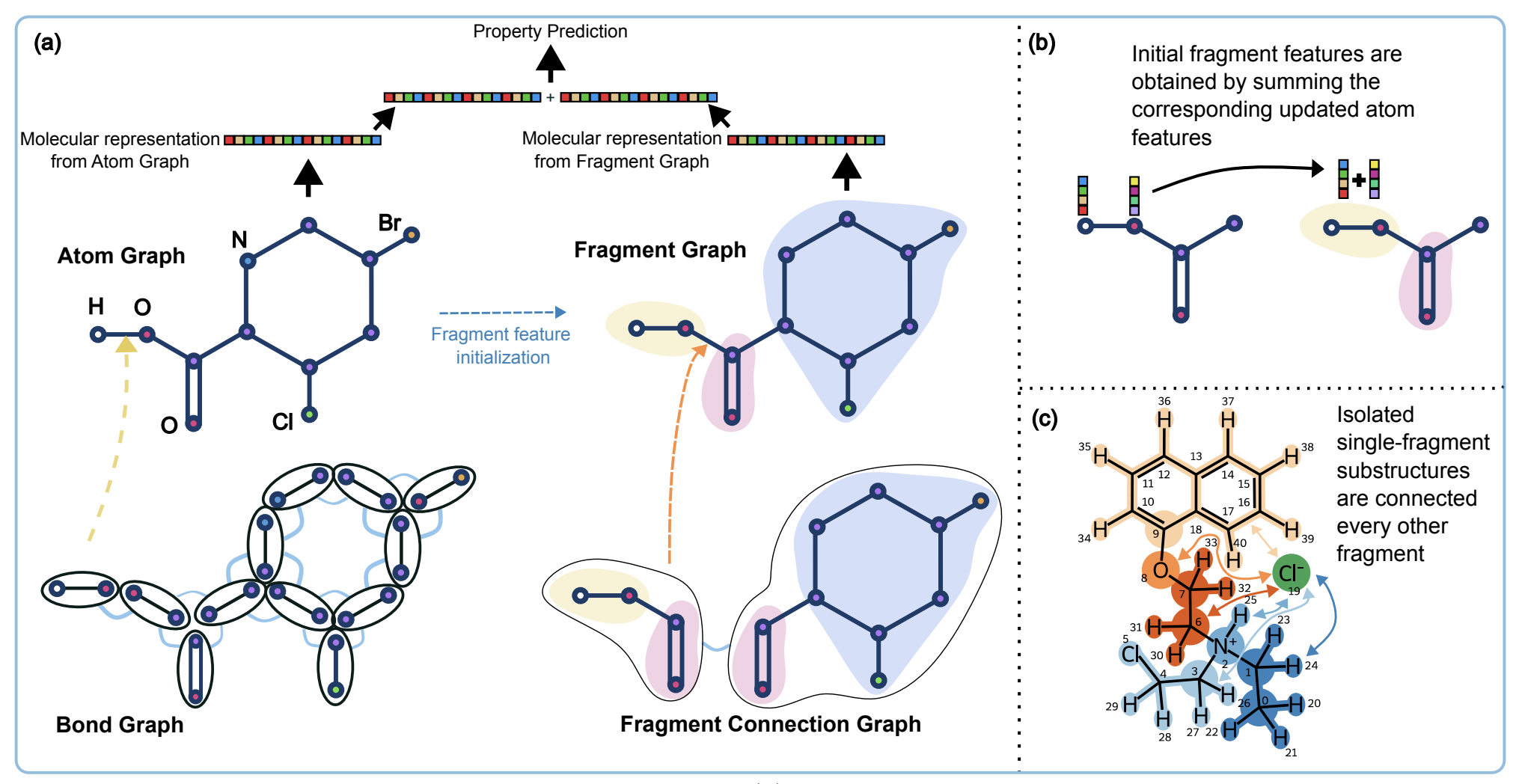
FragNet is an interpretable Graph Neural Network model. FragNet allows for the identification of key atoms, bonds, molecular fragments, and the connections between these
fragments that are essential for predicting a specific molecular property. It is especially valuable for understanding
the significance of connections between fragments in molecules with substructures that do not rely on traditional covalent bonds. Fragnet can also
quantify the contribution of fragments the property prediction, allowing the identification of fragments that may improve or degrade a property value.
Impact of Molecular Representations on Deep Learning Model Comparisons in Drug Response Predictions
Deep learning (DL) is key to predicting drug responses in cancer, but its effectiveness is often limited by
inconsistent benchmarks and data sources. To address this, we introduce the CoMParison workflow for Cross Validation (CMP-CV),
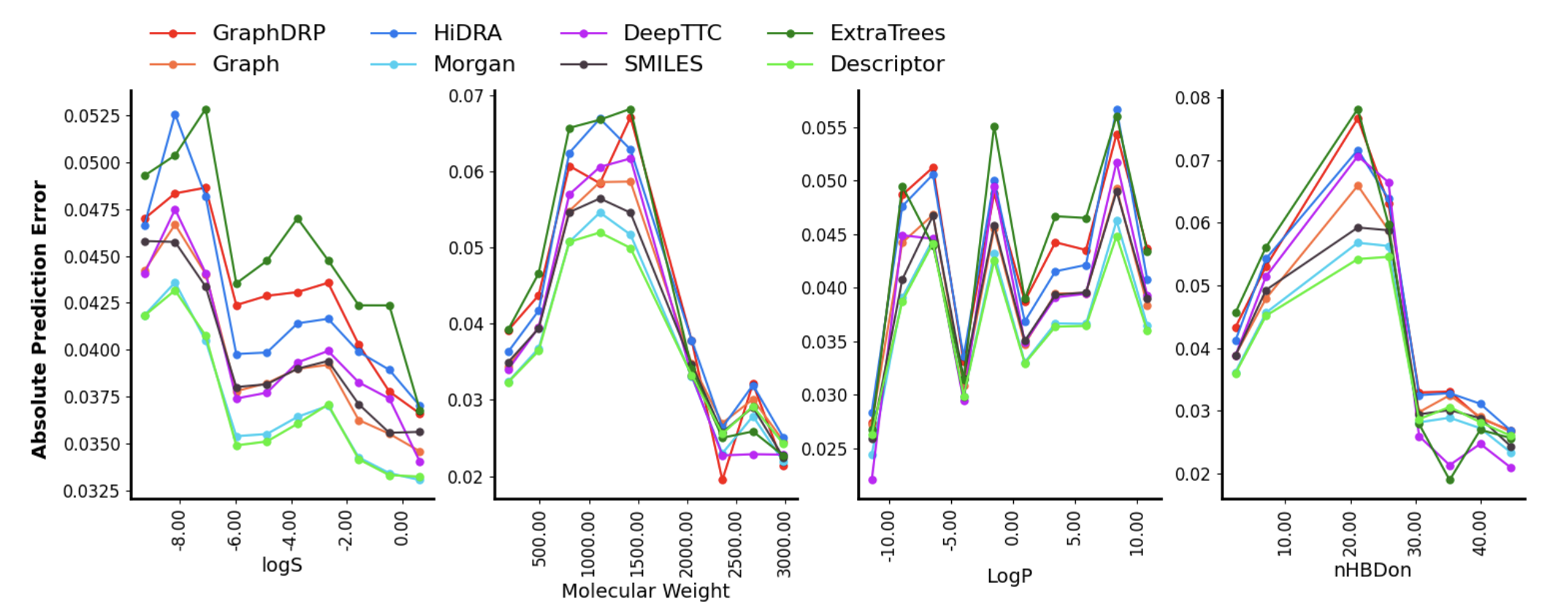
an automated framework for training models with user-defined parameters and metrics. We benchmark several drug representations—graphs,
molecular descriptors, fingerprints, and SMILES—to evaluate their predictive performance. Our results show that molecular descriptors and
Morgan fingerprints perform slightly better overall, though performance varies across different regions of the descriptor space.
This underscores the importance of domain-specific model comparisons. Our work is part of the
CANcer Distributed Learning Environment (CANDLE), advancing model comparison for more effective drug response prediction.
Outlier-Based Domain of Applicability Identification for Materials Property Prediction Models
Machine learning models are widely used for predicting material properties, but their practical application is
limited by uncertainty in their performance on unseen materials. Since predictions depend on the quality of training data,
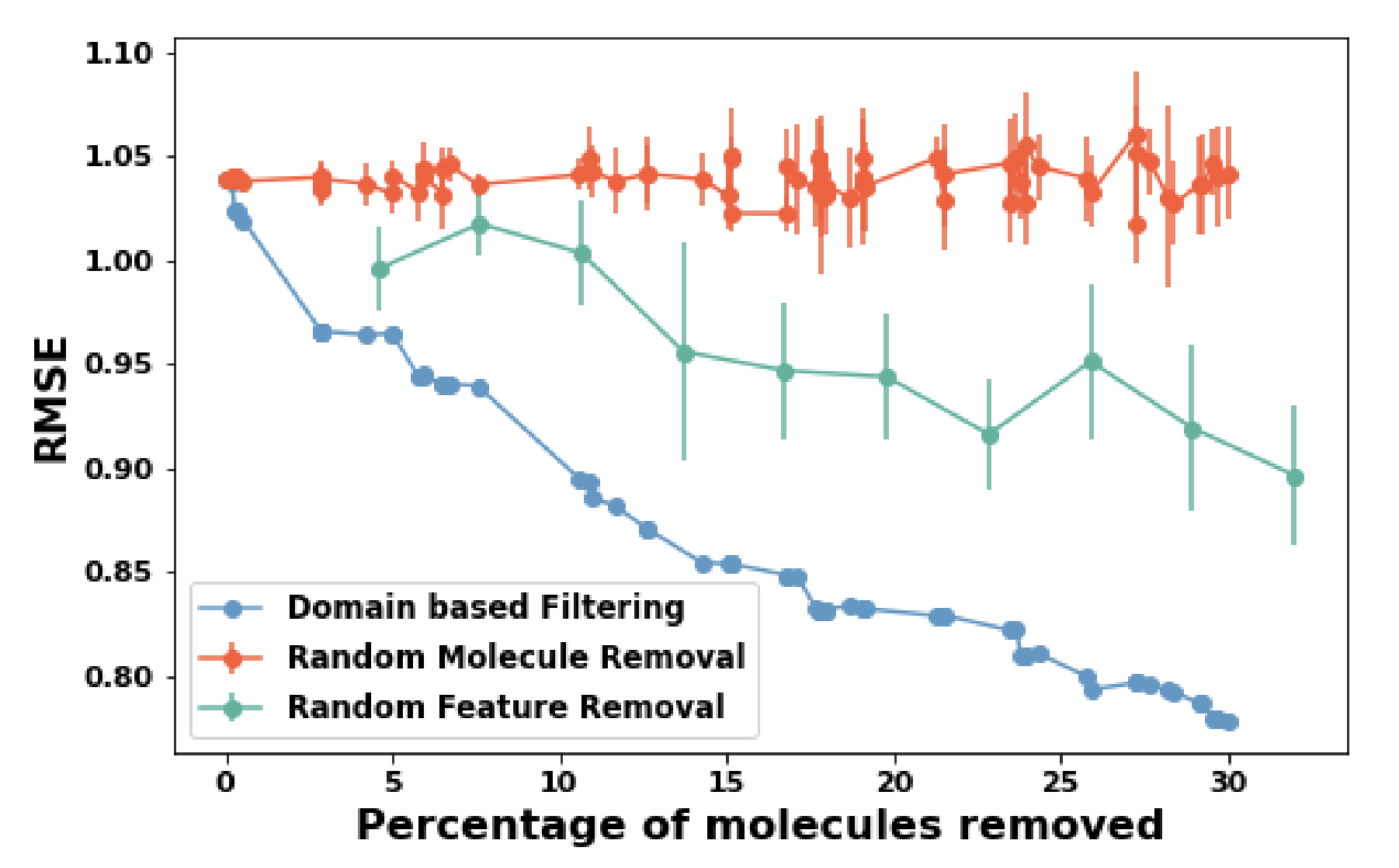
different regions of the material feature space are predicted with varying accuracy. Identifying these regions allows for assessing
the confidence level of predictions, determining when and how to use the model based on accuracy requirements, and improving performance
in error-prone domains. In this work, we propose a method to identify applicable domains within a
large feature space and introduce analysis techniques to better understand these domains and their subdomains.
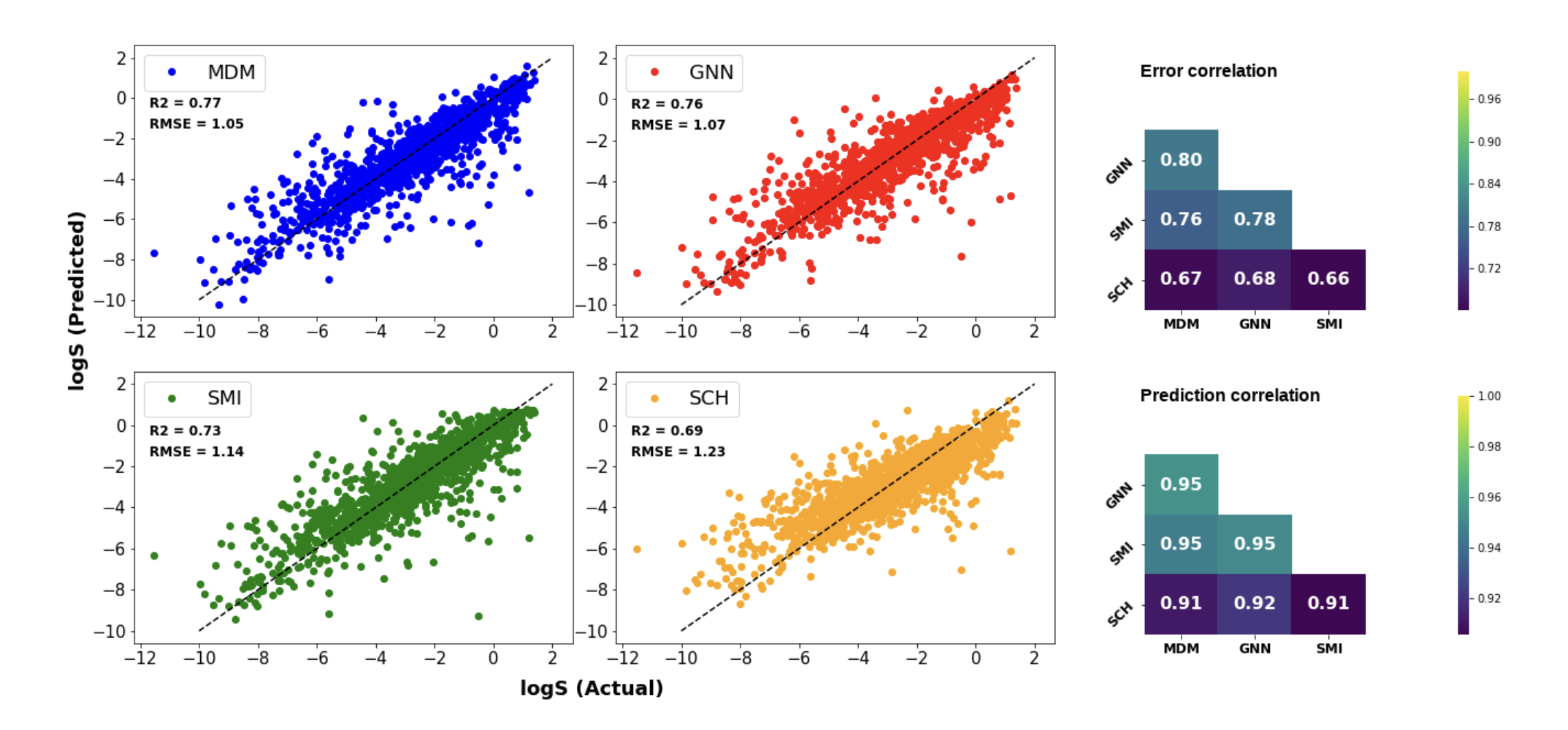
Evaluation of deep learning architectures for aqueous solubility prediction
Accurately predicting aqueous solubility is critical in pharmaceuticals, environmental science,
and energy storage. Despite extensive research, achieving reliable solubility predictions remains challenging.
This study evaluates deep learning methods for solubility prediction, develops a versatile model for organic molecules,
and examines the impact of data, molecular representations, and model architectures on performance. Using the largest
available solubility dataset, we explore molecular descriptors, SMILES, molecular graphs, and 3D atomic coordinates with
four neural network architectures: fully connected networks, recurrent networks, GNNs, and SchNet. Our results show that
molecular descriptors perform best, with GNNs also yielding strong results. We also analyze error patterns, identify key molecular features for prediction,
and assess how data availability affects model performance through transfer learning and dataset size studies.